How to Install LAMMPS on Ubuntu: A Comprehensive Tutorial
Learn the step-by-step process to install LAMMPS on Ubuntu. This guide provides detailed instructions on how to set up LAMMPS for your molecular dynamics simulations.
MolecularMindset
3/11/20244 min read
Tutorial Video Available Here:

LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator) is a powerful molecular dynamics program used by physicists, chemists, materials scientists, and engineers to model particles in various simulations. This tutorial will guide you through the detailed process of installing LAMMPS on Ubuntu, ensuring you have a smooth and successful setup to begin your scientific exploration.
Prerequisites
Before diving into the installation process, ensure your Ubuntu system meets the following requirements:
An updated version of Ubuntu (18.04 LTS or newer recommended).
Sudo privileges to install necessary packages.
Basic familiarity with terminal commands.
Getting Started with the Installation
Firstly, update your package repository to ensure you have access to the latest software versions:
Next, install essential compiler and build tools:
Installing Pre-requisites
MPI (Message Passing Interface) Installation
LAMMPS requires MPI for parallel computation. Install MPI using:
FFTW (Fast Fourier Transform in the West) Installation
FFTW is necessary for certain LAMMPS packages:
Other Optional Packages
Other dependencies may be required depending on the simulations you plan to run. Refer to the LAMMPS documentation for a comprehensive list.
Downloading LAMMPS
You can choose between the stable and development versions of LAMMPS from the official LAMMPS website and download the version suited to your needs.
Configure LAMMPS for compilation using CMake. Additional packages can be added using the -D PKG_NAME=yes flag. Added packages are required for specific types of simulations.
Additional information: https://docs.lammps.org/Packages_details.html
Compile LAMMPS using your available number of processors (nproc)
Post-installation Steps
Set up the environment by adding LAMMPS to your path (adjust the path according to where LAMMPS is installed):
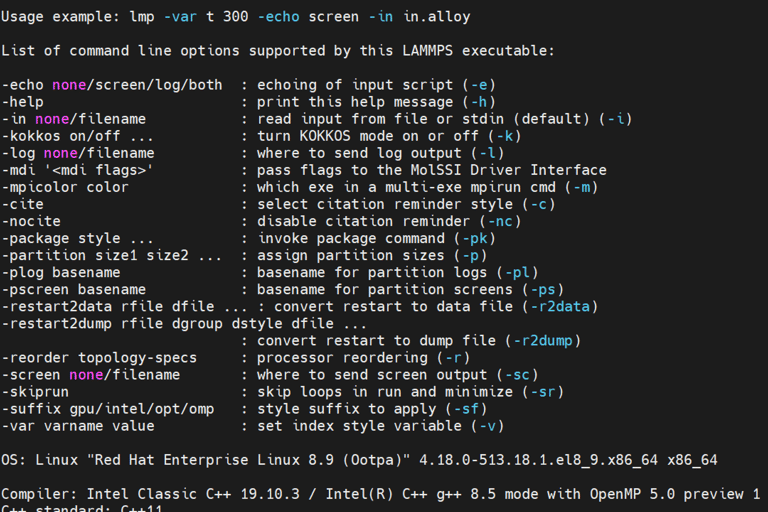
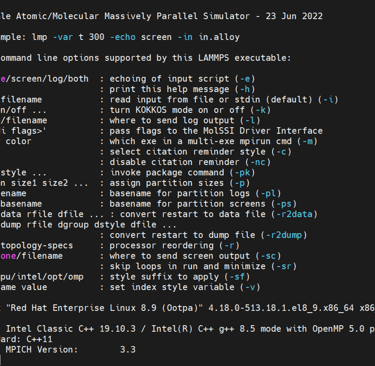
Verify Installation using the helper flag. Detailed information about LAMMPS will appear is sucessfully installed.
Downloading VMD
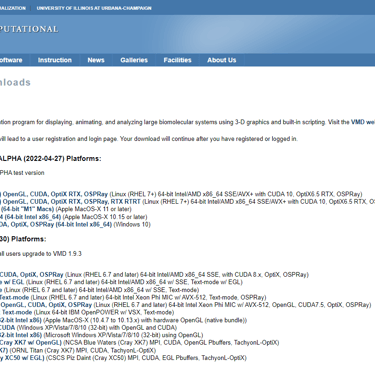
Download VMD from the official website. You may need to register for an account. Choose the version appropriate for your needs and system architecture.
Link: https://www.ks.uiuc.edu/Development/Download/download.cgi?PackageName=VMD

Preparing for Installation
Before installing VMD, install necessary libraries and dependencies:
Installation Process
After downloading, extract the VMD archive and navigate to the directory:
Run the installation script (configure.sh) then do to the src file and install VMD
VMD is automatically added to your PATH so you can run vmd from anywhere in the terminal. Test the installation running VMD.
Run simple molecular dynamics, visualize, and analyze.
Go to this tutorial's repository to obtain sample files for a ethanol simulation. The YouTube video and the repository provide additional instructions on how to run this simulation and visualize it.
Repository: https://github.com/MolecularMindset/Lammps_VMD_Intro
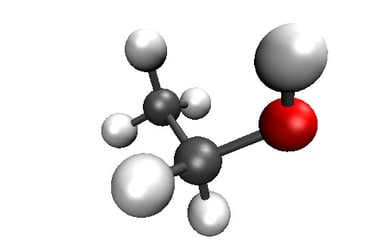
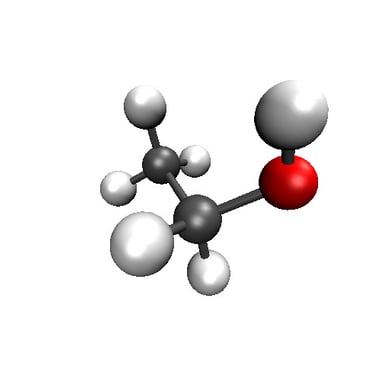
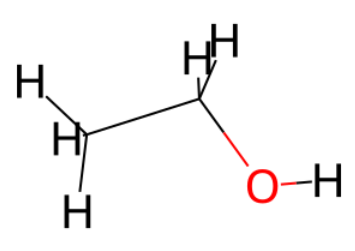
2D visualization of Ethanol molecule with RDkit
3D visualization of Ethanol molecule with VMD
Troubleshooting Common Installation Issues
Encountering issues? Common problems include missing dependencies and compilation errors. Refer back to the prerequisites and ensure all necessary software is installed.
LAMMPS Resources and Community
For further guidance, the LAMMPS documentation is an invaluable resource. Additionally, the LAMMPS community forums provide support and insights from experienced users.
VMD Resources and Community
Dive deeper into VMD's capabilities through the official documentation and join forums for expert advice and community support.
Follow us and Subscribe to keep up with the latest news and Tutorials
Future tutorials include:
How to simulate any organics Solvent using LAMMPS or GROMACS
Pro tips for VMD analysis and visualizations
Introduction to Machine Learning for Molecular Dynamics
FAQs
Can I install LAMMPS on a version of Ubuntu older than 18.04? While possible, older versions may lack the necessary repositories and dependencies for a smooth installation.
How do I update LAMMPS to a newer version? Download the latest version and follow the installation process again. It's advisable to remove the older version first.
What should I do if I encounter a runtime error? Ensure all dependencies are correctly installed and that your simulation script is error-free. Consult the LAMMPS documentation and community forums for specific error messages.
Can I use LAMMPS on a single-core machine? Yes, LAMMPS can run on a single core, but parallel computing environments are recommended for efficiency.
How can I contribute to LAMMPS development? Contributions are welcome! Check the LAMMPS GitHub repository for how to contribute through code, documentation, or community support.
Can VMD run on older versions of Ubuntu? While possible, newer versions provide a smoother experience and better support.
How can I update VMD? Download the latest version from the official website and follow the installation process again.
What if VMD doesn't launch after installation? Ensure all dependencies are installed and your graphics drivers are up to date. Consult the VMD forums for specific issues.
Can VMD visualize data from other molecular dynamics software? Yes, VMD supports a wide range of data formats from various simulation software.
How do I contribute to VMD development or suggest features? The VMD community is active and welcomes contributions. Check the VMD website for how to get involved.
Compiling LAMMPS
Unpack the downloaded tarball and navigate to the LAMMPS directory: